
罕病病友人數少、缺乏藥物,是照護者與醫師心中共同的痛。臺大醫院基因醫學部費時16年,投入藥物研發和臨床試驗,完成全球首例AADC(芳香族L-胺基酸類脫羧基酶)缺乏症的基因治療,這項基因治療藥物更在近年獲得歐洲藥品管理局(EMA)上市許可,讓罕病的基因治療看到一絲曙光。
正常的孩童在3個月時通常會有頭控能力、6個月可以坐起來,看著孩子成長,令父母感到歡喜。然而,台灣每3~5萬人會有1人罹患先天代謝異常疾病AADC缺乏症,以全球統計來說,每3個病例就有1例來自台灣,帶有此基因缺陷的人數少,且集中在台灣與東南亞。
這群孩童因缺乏酵素AADC,難以合成神經傳導物質多巴胺,腎上腺素與正腎上腺素也不足,臨床表現為低張力、躺著無法動彈、情緒不穩,出現動眼危象(雙眼上翻或眼睛偏斜某方向)、自律神經系統功能失調等。有的人症狀會持續6~8小時或每2~3天就發作一次,不利於餵食和成長;以往無藥可醫,病童很難活超過5歲。
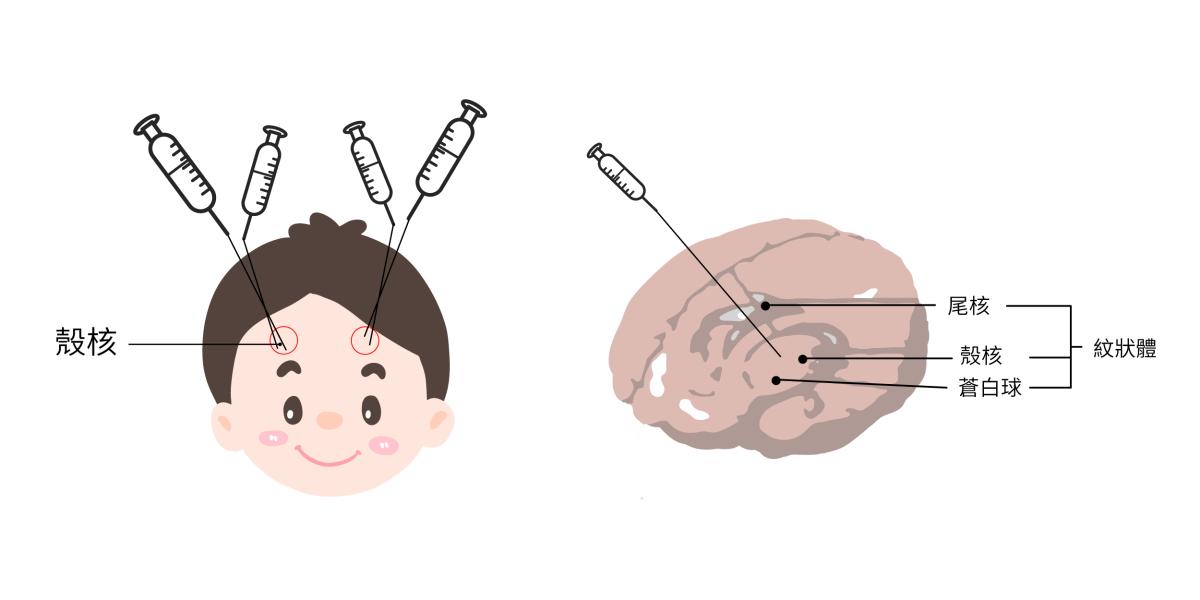
以病毒為載體包裹AADC基因注射於顱骨內,最小能治2歲以下幼童
財團法人罕病基金會育成AADC缺乏症病友2007年聯誼會上,剛好有一位家屬是醫學相關背景,他爬梳資料,發現帕金森氏症有AADC活性喪失、造成多巴胺缺乏的現象,於是與臺大醫學院前教授(現為中國醫藥大學附設醫院精準醫療中心特聘研究員)胡務亮討論,開啟臺大醫院基因醫學部尋找致病機轉,並多次與日本、美國跨科部討論治療的可行性。
罕病基金會日後啟動「罕見疾病臨床試驗計畫補助辦法」,協助本土臨床試驗。2010年臺大醫院獲得衛福部同意,以恩慈療法完成全球首例AADC缺乏症孩童的基因治療。
臺大醫院基因醫學部主治醫師徐瑞聲說明,AADC缺乏症治療是以腺相關病毒為載體,包裹AADC基因後注射,早期是在頭上置入鋼釘再定位;考量孩童的頭圍小、顱骨較軟,現今透過立體定位在孩童頭部精準定出施打位置,以機械手臂將長針注射藥物到腦中基底核的殼核(putamen),因殼核直接受到多巴胺製造缺失影響而成為基因治療的標的,最小已能治療小於2歲的病童。
40%孩童治療後可行走,及早接受基因治療效果愈好
臺大醫院基因醫學部主任簡穎秀說,病童年紀愈小時接受治療,效果愈好。通常AADC缺乏症病童平均被診斷的年齡是3歲,但是請家長回顧,孩童可能出生2、3個月就有症狀。先前臺大醫院比較過1歲半和8歲接受治療的病童表現,1歲半的病童治療後可以走路和上學,甚至能說一些簡單的話;另也觀察到,本來症狀就比較輕微、營養底子好的病童,及早接受治療,也較有潛力改善病況。
接受基因治療後,有語言能力的孩童其實不多,但從完全不能動,到可行走的孩童(含獨立或需要幫忙)占40%,和完全無方法治療已有顯著的差別。
有了基因治療,更促使團隊開發篩檢工具3-OMD血片,臺大醫院推動AADC缺乏症新生兒篩檢先驅計畫,全台99.5%的新生兒都有做此篩檢。簡穎秀說,3-OMD是多巴胺代謝途中的一項指標,AADC缺乏症的病人驗出來的指標特別高,醫院會為孩童進一步安排基因檢測。
如今,臺大醫院神經科或新生兒科醫師對於AADC缺乏症臨床表徵也更了解,能與腦性麻痺區分。如果經基因檢測確診為AADC缺乏症,醫師就會給予藥物治療,並向家屬解釋接下來可能遇到的問題,如孩童吃奶的狀況不順、體重太輕等,請物理治療師或營養師介入,讓孩童的發展盡量不受到病情影響。
台大與美、英、歐合作技術轉移
AADC缺乏症病童經臺大醫院的基因治療後,個案5年存活率達93.5%,吸引美國生醫公司Agilis Biotherapeutics主動聯繫,成功簽署產學合作技術轉移計畫。後來Agilis Biotherapeutics被美國藥廠PTC Therapeutics併購,在法國、德國推動「擴大取得」(Expanded Access Program,簡稱EAP) ,並在美國展開臨床試驗,嘉惠更多病人,而英國目前已核准該藥物上市。
2022年,基因治療藥物Upstaza™(學名Eladocagene exuparvovec)獲得歐洲藥品管理局授予上市許可,成為全球唯一通過歐盟認證治療AADC缺乏症的腦部基因治療藥物。
臺大醫院基因醫學部在AADC缺乏症的基因治療成果傲視全球,包含治療年度最早、治療病例數最多、能治療最小年紀的孩童,注射的部位也最淺和安全,獲得兒科聖經級教科書《納爾遜簡明兒科醫學(Nelson Textbook of Pediatrics, 21st Edition)》引用。
國際上也出現追隨的競爭者,如美國加州大學舊金山分校(UCSF)在臨床試驗第一期有7位AADC缺乏症病例參與。臺大醫院基因醫學部除了協助東南亞國家新生兒血片篩檢,也發表治療照護指引、爭取藥物納保,持續發揮國際影響力,為更多罕病兒翻轉人生。
編按:團隊為第25屆(2023年)國家生技醫療品質獎/醫療院所類特色醫療組金獎,文中所提及之職稱,皆為受訪當時之職務。




